|
Objective and Overview
The objective of this tutorial is to
demonstrate the use of simplfied representations of the
protein chain, in this case Go-type models, to explore the unfolding of proteins via single molecule pulling experiments.
This example utilizes the MMTSB Go Model Server
to create a Go-type model from the relevant pdb file (1QJO.pdb)
and then carries out pulling experiments, using the CHARMM afm module, via constant speed pulling. The protein is
streched from two different points.
First streching from the N- and C-termini is examined. Next the "experiments" are
run by connecting the pulling point to the N-terminus and at residue 41, midway through the structure.
Very different streching profiles arise from these two different experiments. Moreover, the results from this
simple model correlate very well with experiments and earlier calculations by Brockwell et al.
[Brockwell et al., NSB, 10, 731 (2003)]
The Go model we employ is a minimalist representation of the protein where each amino acid residue is described by
one "bead," located at the α-carbon position. The mass of each bead is taken to be the mass of the amino acid
residue that the bead represents. The beads are connected via "virtual bonds" along the protein backbone.
See Karanicolas and Brooks,
Protein Science (2002), 11:2351-2361, for details of the model and its use in studying protein folding.
|
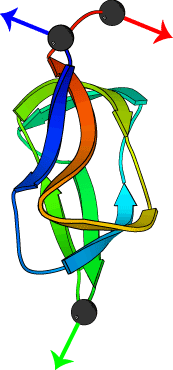
Pulling geometry defines the mechanical resistance of a
beta-sheet protein
|
|
This example uses CHARMM and the input file
pull_GoModel.inp together with the files that came from the Go Model Server noted above, for the pdb fle 1QJO these are:
GO_1qjo.Qdetails, GO_1qjo.Qlist, GO_1qjo.param, GO_1qjo.pdb, GO_1qjo.seq, GO_1qjo.top.
You can download all of the files as a single bundle here (gzipped tar file). The CHARMM
input script to run this example will execute in a reasonable amount of time (less than 20 minutes on 3GHz Mac
for 2 trials).
Running it on the CTBP Linux cluster will be faster if you wish. To run this script you execute the commands:
$CHARMMEXEC ntrials=1 last=80 < pull_GoModel.inp > pull_1_80.out
$CHARMMEXEC ntrials=1 last=41 < pull_GoModel.inp > pull_1_41.out
The first command will run a pulling simulation with the pulling force applied to the first and last (residue 80) residues.
The second will pull on the first and 41st residue. Multiple pulling simulations can be run using a larger value of
ntrials.
In the input file the number of steps is set at 100000, which is not enough to fully unfold the protein when
pulled from the ends.
Feel free to change Nsteps to extend the simulation.
The input file (pull_GoModel.inp) for this example is given below:
|
|
* This CHARMM input script demonstrates the use of constant force (cf)
* and constant speed (steered-molecular dynamics, smd) pulling
* experiments on an all beta protein E2lip3 (PDB: 1qjo.pdb).
* The experiments explore the differences in mechanical stability and
* mechanical stress induced unfolding of this protein as a function of
* where the attachment is made. The calculations utilize the sequence-dependent
*
Go-model of Karanicolas and Brooks and mirror the earlier study of
* Brockwell et al. (NSB, 10, 731-738 (2003)).
* This input script was developed following a template provide by
* Emanuele Paci from the University of Zurich in July 2004.
* The only input to this script is whether one is running an constant-force
* pulling calculation (Method = cf) or a constant speed pulling calculation
* (Method = smd) (the default is smd), where the cantilever is attached
* (Last = 80/41), and the number of pulling trials (Ntrials = x).
**
**
* Required files: go_[at]prot.top, go_[at]prot.param, go_[at]prot.seq,
* go_[at]prot.pdb
* where these files are from the MMTSB Go Model Server and the charmm
* variable prot (in [at]prot) is defined as prot = 1qjo
**
**
* Usage: charmm Method=[cf/smd] Last=[80/41] NTrials=[x] < pull_GoModel.inp > pull_GoModel.out
**
**
*
Prepared by C.L. Brooks III, The Scripps Research Institute, July, 2004.
*
if @?Method eq 0 set Method = smd !set default method to smd
if @?Last eq 0 set Last = 80 !default is to pull from end
if @?Ntrials eq 0 set Ntrials = 1 !default number of runs is 1
SET SegName = prot ! Name of the domain
SET prot = 1qjo
SET Temp = 300 ! temperature for heatbath during pulling
SET TimeStep = 0.010 ! with Go models we can use a large timestep
SET Bias = 100 ! constant force in pN
SET Nsteps = 100000 ! Number of timesteps
SET First = 1
Calc Lmax = (@Last - @First) * 3.8 * 1.4 ! approximate maximum length of peptide
if Method eq smd SET Speed = 0.1 ! pulling speed with SMD (Angstroom/ps)
eten on ! Turn on the special interaction term for Go models
! Stream in the Go model parameters
STREAM "GO_@PROT.top"
STREAM "GO_@PROT.param"
STREAM "GO_@PROT.seq"
set cnt = 1
label dopull
! Set up a random seed
system "echo 'set iseed = ' `date +%H%M%S` > seed.stream"
stream seed.stream
system "rm seed.stream"
calc iseed = 2* @iseed + 1
! Read in the protein coordinates
OPEN UNIT 1 READ FORM NAME "GO_@PROT.pdb"
READ COOR PDB UNIT 1
CLOSE UNIT 1
ENERGY
SCALAR FBETA SET 0.1 SELECT all END ! Use friction coefficient to couple to Langevin
! heatbath.
SHAKE BOND TOL 1.0E-6 PARAM ! Use constraint to fix bonds
COOR DIST SELE (ATOM @SegName @First CA) END SELE (ATOM @SegName @Last CA) END
DEFINE nc SELE ((ATOM @SegName @First CA) .OR. (ATOM @SegName @Last CA)) END
OPEN WRITE FORMATTED UNIT 32 NAME @Method@Bias.@First-@Last_@cnt.rst
OPEN WRITE UNFORMATT UNIT 34 NAME @Method@Bias.@First-@Last_@cnt.dcd
OPEN WRITE FORMATT UNIT 44 name @Method@Bias.@First-@Last_@cnt.juj
! { Pull with constant force }
if Method eq cf AFM @Method IUNJUJ 44 ALPHA @Bias SELE nc END XIMAX @Lmax
! { Pull with steered molecular dynamics }
if Method eq smd AFM @Method IUNJUJ 44 ALPHA @Bias BETA @Speed SELE nc END XIMAX @Lmax
DYNAMICS LEAP LANGEVIN START TIMESTEP @Timestep -
NSTEP @Nsteps NPRINT 100 IPRFRQ 50000 -
FIRSTT @Temp FINALT @Temp TWINDL -10.0 TWINDH 10.0 -
ICHECW 1 IEQFRQ 500 IASORS 1 IASVEL 1 -
NSAVC 1000 NSAVV 0 IUNVEL -1 -
IUNREA 31 IUNWRI 32 KUNIT -1 IUNCRD 34 -
INBFRQ 0 ILBFRQ 0 TBATH @Temp ECHECK 9999999999.0 ISEED @iseed
OPEN UNIT 1 WRITE FORM NAME @Method@Bias.@First-@Last_@cnt.pdb
WRITE COOR PDB UNIT 1
CLOSE UNIT 1
afm reset ! reset afm module for next trip through code
incr cnt by 1
if cnt le @Ntrials goto dopull
STOP
Analysis
- VMD View trajectory of pulling experiment using vmd
- Load pdb file to vmd (GO_1qjo.pdb)
- View as "trace"
- Load trajectory file (*.trj or *.dcd files) and replay movie
- XMGRACE Graph pulling force vs. distance Data is in *.juj, see charmm doc afm.doc. We need cols 2 and 4
- Get xmgrace for your MACget grace
- Filter columns needed: % awk '{print $2,$4}' smd100.1-80_1.juj > fvsd.dat
- start xmgrace, menu: Data/Import/Ascii, read in fvsd.dat
- Use a running average to level out spikes: menu Data/Transformations/RunningAverages
- specify 50 for length of average; Accept; Close
- Delete the old data set. menu Edit/Data sets, click on G0.S0 menu edit/kill ; OK; Close
- EXCEL
- Start excel, File/Open
- Find your directory, Enable: All Documents, Click on the .juj file (smd100.1-80_1.juj)
- Next; Next; Finish
- Select Column B and D (Click "B", then Apple click "D"), Click above on "Chart Wizard"
- X-Y Scatter; then lower right selection; next; next; finish
- Click on noisy line in graph; then Control-Click; Trendline; Moving Average; 50 period;OK
- Double click on noisy line; None on left line; OK Whew, we are done.
|
|
Results of multiple pulling "experiments".
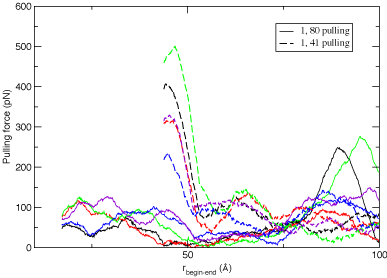
|
|
Calculations carried out for multiple trials yield "traces" of the force required to strech the
protein versus the distance from one point
of connection to the next illustrate the qualitative difference that arises in the mechanical
unfolding of a protein when it is streched from different positions. The results from these calculations are shown in
the figure below, and illustrate that the protein unfolds with less force when pulled from the ends versus the force
required when pulling from the center. What are the features of the protein topology
that contribute to this behavior?
|
|
Representative streching trajectories
|
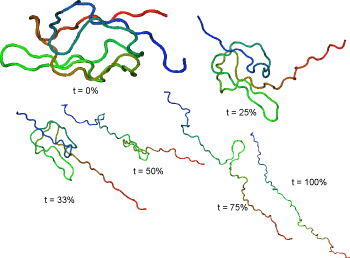
Streching from N- and C-terminal ends
|
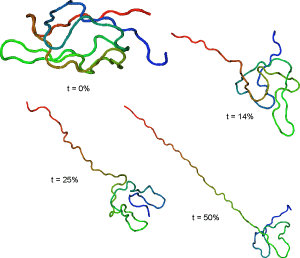
Streching from N-terminus and resdue 41
|
Acknowledgement: This tutorial example was contributed by Emanuele Paci from the University of Zurich
Suggested exercises and simulation experiments
- Pulling experiments on same protein
- Use different first and last residues. Either use first= and last= args to charmm with the pull script or
edit the AFM command (for more information, see CHARMM afm.doc).
- Use different pulling speed or try constant force method. Edit the script or use Method=cf or edit the
AFM command in the script
- Download another pdb file such as protein L (2PTL) or Protein G (1PGB)
- Get the pdb from PDB repository
- Submit the pdb to MMTSB Go Model Server
- Get the files from your email and try them out with CHARMM and the pull input script (use prot= arg for charmm execution)
INSTALLING XMGRACE ON MAC
- grace-macosx.tar.gz
- Uncompress, untar: tar zxvf grace-macosx.tar.gz
- Set up your environment: cd grace; export GRACE_HOME=`pwd`
- alias the command: alias xmgrace=$GRACE_HOME/bin/xmgrace
|

